又一上市创新产品,审评报告全文公开
3月22日,国家药品监督管理局医疗器械技术审评中心发布《腹主动脉覆膜支架及输送系统(CQZ1800050)》,该产品由自由扩展的覆膜支架与相应的输送系统组成,覆膜支架预装载在输送系统中。以下是详细内容:
基本信息
一、申请人名称
上海微创心脉医疗科技股份有限公司
二、申请人住所
上海市浦东新区康新公路3399弄1号
三、生产地址
上海市浦东新区康新公路3399弄1号,芙蓉花路388号3号楼一层和三层、4号楼二层和三层
产品审评摘要
一、产品概述
(一)产品结构及组成
该产品由覆膜支架与相应的输送系统组成,覆膜支架预装载在输送系统中。覆膜支架包括主体覆膜支架、分支覆膜支架、CUFF覆膜支架,均由聚酯膜通过聚酯缝合线与多个自扩张的镍钛合金支架段缝合,带有铂铱合金和钽显影点。镍钛合金支架段由镍钛丝编制而成,主体覆膜支架裸段有倒钩结构。输送系统用于实现覆膜支架在人体内的输送和释放,主要由锥形头、内管、外管、手柄、输液管等零部件组成。
产品经环氧乙烷灭菌,一次性使用。
图1 覆膜支架示意图
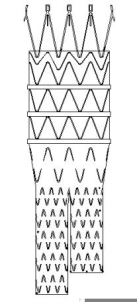
a)主体覆膜支架
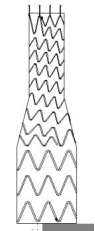
b)分支覆膜支架
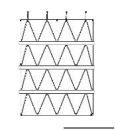
c)CUFF覆膜支架
图2产品示意图
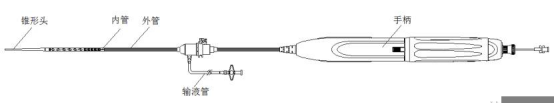
(二)产品适用范围
该产品适用于近端瘤颈长度≥15mm的腹主动脉瘤。
(三)型号/规格
产品的型号规格参见表1。
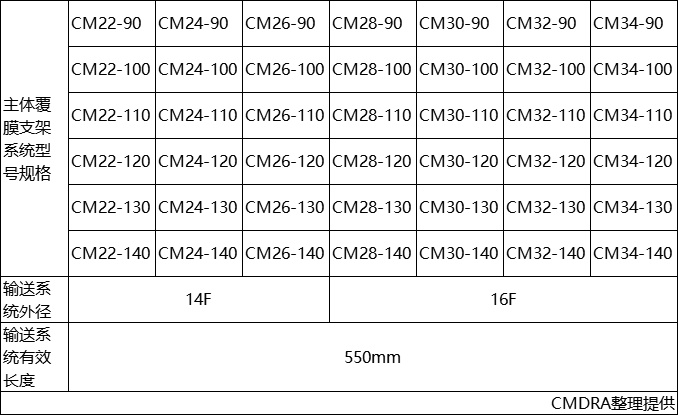
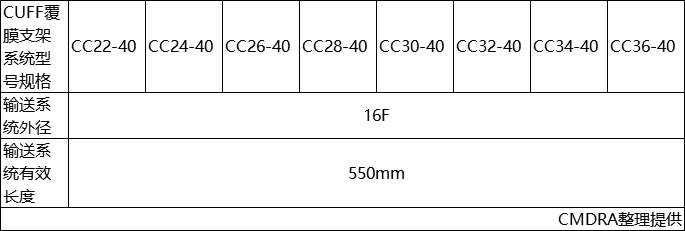
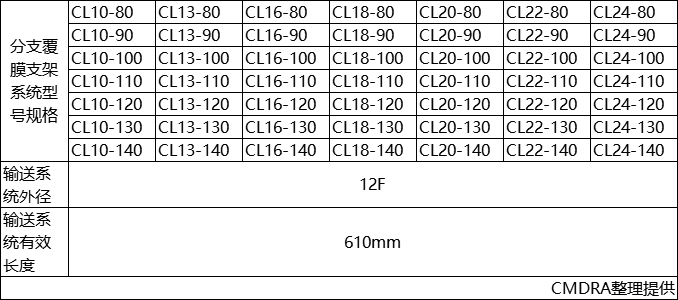
表1型号规格表
产品型号规格代码示例如下:
①CM22-90
其中
CM代表主体覆膜支架系统
22代表主体覆膜支架近端直径
90代表主体覆膜支架同侧覆膜长度
②CC22-40
其中
CC代表 CUFF覆膜支架系统
22代表CUFF覆膜支架直径
40代表CUFF覆膜支架覆膜长度
③CL13-80
其中
CL代表分支覆膜支架系统
13代表分支覆膜支架远端直径
80代表分支覆膜支架覆膜长度
(四)作用机理
本产品通过特定的输送系统,将由镍钛合金支架和覆膜材料缝合而成的覆膜支架释放在病变位置,支撑在病变血管两端的正常血管壁上,代受血流对病变血管壁的压力,将瘤腔隔绝在血液循环系统之外,使血管瘤壁不再承受血压而避免瘤体破裂。血管外壁因负压而回缩,慢慢恢复正常血管形态,血管内膜细胞渗透覆膜材料中的微孔,最终完全覆盖覆膜支架。
二、临床前研究摘要
(一)产品性能研究
1. 产品技术要求研究
技术要求研究项目如表2所示:
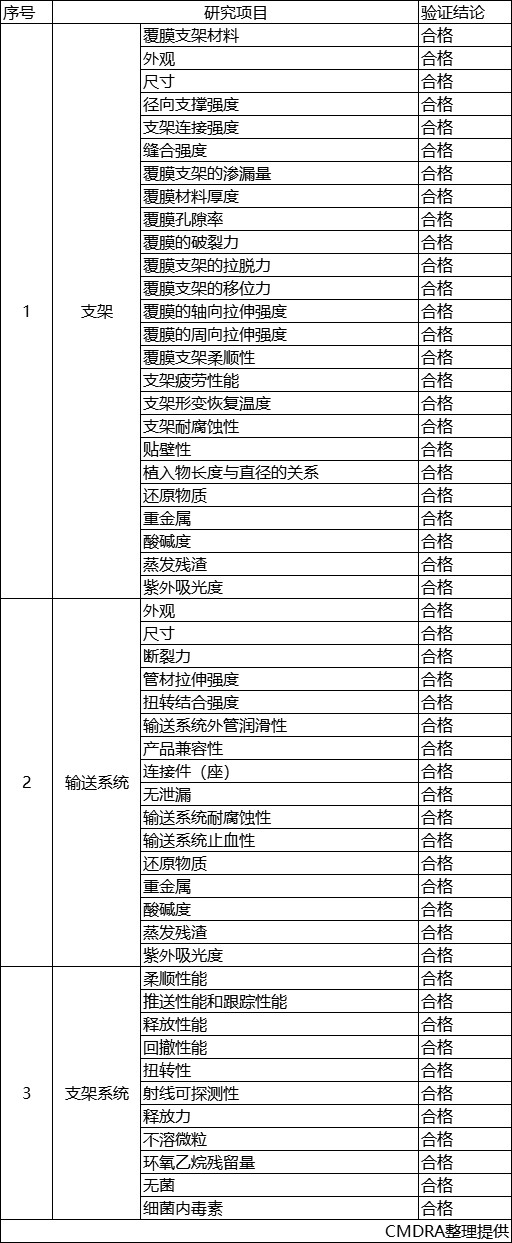
表2技术要求研究摘要
2. 产品性能评价
产品性能评价包括覆膜支架规格与输送系统配对关系、MRI兼容性、尖端构形、水合性、支架系统轮廓、支架系统注水排空性、支架有限元分析等性能验证,并对支架段热处理过程、支架段焊接过程、制作外管过程、亲水涂层过程、清洗过程、封口过程等工艺进行了验证,结果表明产品符合设计输入要求。
(二)生物相容性
该产品包含覆膜支架和输送系统两部分,其中支架为植入器械,与循环血液长期接触;输送系统为外部接入器械,与循环血液短期接触。申请人按照GB/T 16886 系列标准对植入器械及外部接入器械分别进行了生物相容性评价,产品的生物相容性风险可接受,具体评价项目详见表3。
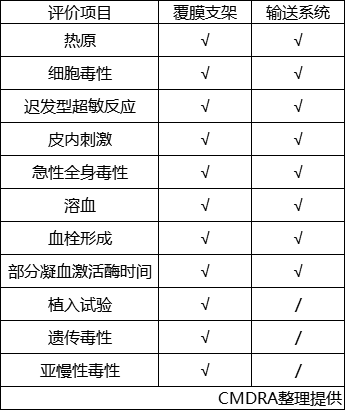
表3生物相容性评价项目表
(三)灭菌
该产品采用环氧乙烷灭菌,无菌状态提供。申请人提供了灭菌过程确认报告,证明无菌保证水平为 10-6。环氧乙烷残留量不大于10μg/g,2-氯乙醇残留水平不超过9mg/件。
(四)产品有效期和包装
该产品有效期为两年。申请人提供了货架有效期验证报告。验证实验为加速老化和实时老化,包括产品稳定性、包装完整性和运输模拟验证。
(五)动物研究
申请人开展了猪模型的动物实验研究以验证产品使用性能及安全性。支架植入即刻、植入后 30天、90天、180天分别进行观察,评价指标包括支架移位情况,支架结构及覆膜的完整性,覆膜支架的通畅率,肾动脉血流是否通畅,生物相容性等。动物实验结果表明,产品达到预期设计要求。
三、临床评价摘要
该临床试验方式为前瞻性、多中心、单组目标值法试验设计,纳入136例受试者验证该产品的安全性及有效性。
临床试验主要终点为 12个月的临床成功率和 30天主要不良事件率。次要终点为器械成功率,主要技术成功率,30天死亡率,12个月器械相关不良事件发生率,6个月/12个月/2-5年二次干预发生率、全因死亡率、动脉瘤相关死亡率、严重不良事件发生率。
共14家临床机构参与该试验,入组受试者 136例,全分析集(FAS)人群为136例,符合方案分析集(PPS)人群为121例。FAS分析集下试验期间12个月无影像证据者按12个月临床失败进行结转后计算的临床成功率为 89%,PPS分析集下12个月临床成功率为95.9%,均大于主要有效终点指标。
FAS分析集下30天MAE发生率为1.5%,PPS分析集下30天MAE发生率为1.7%,均低于主要安全终点指标。结果表明,该临床试验 12个月临床成功率和 30天主要不良事件发生率均满足目标值的要求。详见表4。

表4主要有效及安全终点指标统计分析结果
次要有效性终点指标结果如表5所示:器械成功率为100%;主要技术成功率为 97.1%。

表5次要有效性终点指标结果
次要安全性终点指标结果如表6所示:6个月、12个月随访发生动脉瘤相关的二次干预发生率均为 2.2%;30天死亡率为 0.7%、6个月死亡率为 3.7%、12个月死亡率为 7.4%;6、12个月动脉瘤相关死亡率均为 0.7%。

表6次要安全性终点指标结果
不良事件发生情况如表7所示:6个月发生严重不良事件的患者比例为 31.6%,12个月发生严重不良事件的患者比例为37.5%;12个月随访器械相关的不良事件发生率为17.6%。
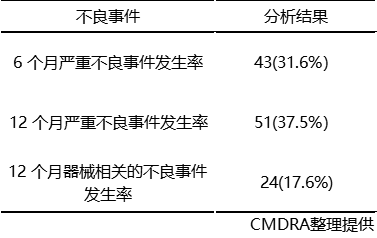
表7不良事件发生情况
该产品在临床应用中通过隔绝动脉瘤,重建血流通道,从而减缓或阻止动脉瘤的增长。但可能伴随动脉壁穿孔或破裂、瘤体破裂、支架移位、内漏等风险。根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品为患者带来的受益大于风险。
四、风险分析及说明书提示
参照《YY/T 0316-2016医疗器械风险管理对医疗器械的应用》,对该产品进行风险分析。对目前已知及可预测风险采取了风险控制措施,经综合评价,在目前认知水平上,认为该产品上市带来的获益/受益大于风险。为保证用械安全,需在说明书中提示以下信息:
(一)明确的产品适用范围
(二)警示及注意事项
1.使用前请仔细阅读使用说明书,以避免不良后果。
2.使用该产品前,医师需经过相应的专业培训,对大动脉支架术的原理、临床应用、并发症、副作用及危害有清楚的了解;
3.本产品仅供一次性使用;
4.拆封前若发现包装破损,请勿使用。
(三)禁忌症
1.动脉瘤附近存在严重狭窄,或者钙化,容易导致覆膜支架难以贴壁者;
2.髂动脉或股动脉严重狭窄、扭曲,无法提供有效的产品进入途径者;
3.破裂性动脉瘤或急性动脉瘤的患者;
4.感染性动脉瘤的患者;
5.对造影剂过敏或肾功能不全而不能耐受造影剂者;
6.患者存在严重凝血功能障碍,易增加术后出血者;
7.瘤颈弯曲角度>60°,远端无足够锚定区,或者容易导致覆膜支架难以贴壁者;
8.患者重要脏器的供血分支血管起于瘤腔;
9.患者并存其他疾病,预期寿命不超过一年者;
10.患有结缔组织病,如马凡氏综合症患者;
11.未成年人及孕妇;
12.对镍钛合金过敏者;
13.其他不适宜放置覆膜支架者。
综合评价意见
本申报产品属于创新医疗器械特别审批项目,编号201600189。申请人的注册申报资料符合现行要求,依据《医疗器械监督管理条例》(国务院令第 680号)、《医疗器械注册管理办法》(国家食品药品管理总局令 2014年第4号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册。
;){kind=link}